{kind=link}
{kind=link}
Integrating PepGM and Unipept for probability-based taxonomic inference of metaproteomic samples
Table of Contents
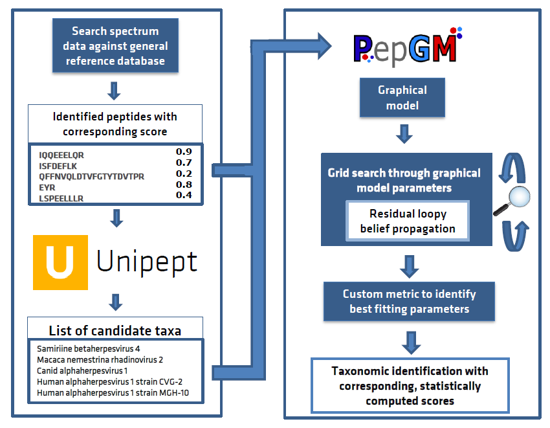
Introducing the Peptonizer2000 - a tool that combines the capabilities of Unipept and PepGM to analyze metaproteomic mass spectrometry-based samples. Originally designed for taxonomic inference of viral mass spectrometry-based samples, we've extended PepGM's functionality to analyze metaproteomic samples by retrieving taxonomic information from the Unipept database.
PepGM is a probabilistic graphical model developed by Tanja Holstein et al. that uses belief propagation to infer the taxonomic origin of peptides and taxa in viral samples. You can learn more about PepGM at GitHub page.
Unipept, on the other hand, is a web-based metaproteomics analysis tool that provides taxonomic information for identified peptides. To make it work seamlessly with PepGM, we've extended Unipept with new functionalities that restrict the taxa queried and provide all potential taxonomic origins of the peptides queried. Check out more information about Unipept here.
With the Peptonizer2000, you can look forward to a comprehensive and streamlined workflow that simplifies the process of identifying peptides and their taxonomic origins in metaproteomic samples.
The Peptonizer2000 workflow is comprised of the following steps:
- Query all identified peptides, provided by the user in a .tsv file, in the Unipept API, and restrict the taxonomic range queried based on any prior knowledge of the sample.
- Assemble the peptide-taxon associations provided by Unipept into a bipartite graph, where peptides and taxa are represented by different nodes, and an edge is drawn between a peptide and a taxon if the peptide is part of the taxon's proteome.
- Transform the bipartite graph into a factor graph using convolution trees and conditional probability table factors (CPD).
- Run the belief propagation algorithm multiple times with different sets of CPD parameters until convergence, to obtain posterior probabilities of candidate taxa.
- Use an empirically deduced metric to determine the ideal graph parameter set.
- Output the top scoring taxa as a results barchart. The results are also available as comma-separated files for further downstream analysis or visualizations.
- A .tsv file of your peptides output from any protoemic peptide search method. The first column should be the peptide, the second column it's score attributed by the search engine. An example is provided in test files.
- A config file with your parameters for the peptonizer2000. A more detailed description of the configuration file can be found below. Additionally, an exemplary config file is provided in this repository.
The actual code that builds the factor graph and executes the Peptonizer algorithm, is implemented in Python and can be found in the peptonizer folder.
In order to run the Peptonizer2000 on your own system, you should install Conda, Mamba and all of its dependencies. Follow the installation instructions step-by-step for an explanation of what you should do.
- Make sure that Conda and Mamba are installed. If these are not yet present on your system, you can follow the instructions on their README.
- Go to the "workflow" directory by executing
cd workflowfrom the terminal. - Run
conda env create -f env.yml(make sure to run this command from the workflow directory) in order to install all dependencies and create a new conda environment (which is named "peptonizer" by default). - Run
mamba install -c conda-forge -c bioconda -n peptonizer snakemaketo install snakemake which is required to run the whole workflow from start-to-finish. - Run
conda activate peptonizerto switch the current Conda environment to the peptonizer environment you created earlier. - Start the peptonizer with the command
snakemake --use-conda --cores 1. If you have sufficient CPU and memory power available to your system, you can increase the amount of cores in order to speed up the workflow.
The Peptonizer2000 relies on a configuration file in yaml format to set up the workflow.
An example configuration file is provided in config/config.yaml.
Do not change the config file location.
Directory parameters
- data_dir: relative path to output files
- input_file: relative path to input .tsv
- log_dir: relative path to log directory
Analysis specific parameter
- taxa_in_graph: # of inferred taxa that appear in the barplot that is created of the results csv
- taxa_in_plot: number of taxa reported in bar plot
- alpha: grid search increments for alpha (list)
- beta: grid search increments for beta (list)
- prior: grid search increments for prior (list)
- regularized: boolean. If True, the probability for the number of parents taxa of a peptide is regularized to be harmonically decreasing with the number of parents
UniPept query parameters
- taxon_rank: rank at which results will be reported
- taxon_query: taxa comprised in the UniPept query. If querying all of Unipept, use 1 (list)
All Peptonizer2000 output files are saved into the results folder and include the following:
Main results:
- peptonizer_results.csv: table with values ID, score, type (contains all taxids under 'ID' and all probabilities under 'score'
- peptonizer_results.png: bar plot of the peptonizer results showing the scores for the #'taxa_in_plot' (see config parameters) highest scoring taxa
Additional files:
- Intermediate results folders sorted by their prior value for all possible grid search parameter combinations
- taxa_weights_dataframe.csv: csv file of all taxids that had at least one peptide map to them and their weight
- pepgm_graph.graphml: graphml file of the graphical model (without convolution tree factors). Useful to visualize the graph structure and peptide-taxon connections
- sequence_scores_dataframe.csv: dataframe with petides, taxa and scores used to create the graph
- best_parameter.csv: file with best parameter
- unipept_responses.json: response of unipept queries
- clustered_taxa_weights_datatframe: additional .csv file resulting from the clustering of taxa by peptidome used for rbo
To test the Peptonizer2000 and see if it is set up correctly on your machine, we provide a test file under resources/test_files. This should be dowloaded automatically if you follow the installation instructions above. There are several test files from different metaproteomic samples. These are:
- the samples S03, S05 and S11 of the CAMPI study searched against a sample specific database using X!Tandem and MS2Rescore. The original files are available through PRIDE under PXD023217.
- the sample U1 of uneven communities from a metaproteomic benchmark study by Kleiner searched against a sample specific database. The original files are available through PRIDE under PXD006118
- the sample F07, a fecal sample, of the CAMPI study searched against the integrated gene catalog for the human gut using X!Tandem and MS2Rescore. The original files are available through PRIDE under PXD023217.
To execute a test run of the Peptonizer2000 using the provided files:
- Follow the installation instructions above
- In the config file, make sure to point to the test sample you want to use. By default, this is S03
- Start to peptonize with the command
snakemake --use-conda --cores 1. If you have sufficient CPU and memory power available to your system, you can increase the amount of cores in order to speed up the workflow.
Distributed under the Apache 2.0 License. See LICENSE.txt for more information.
Tanja Holstein - @HolsteinTanja - [email protected]
Pieter Verschaffelt - [email protected]