English | 简体中文
Ilus (/'i:loʊs/) is a lightweight, scalable, handy semi-automated variant calling pipeline generator for Whole-genome sequencing (WGS) and Whole exom sequencing (WES) analysis.
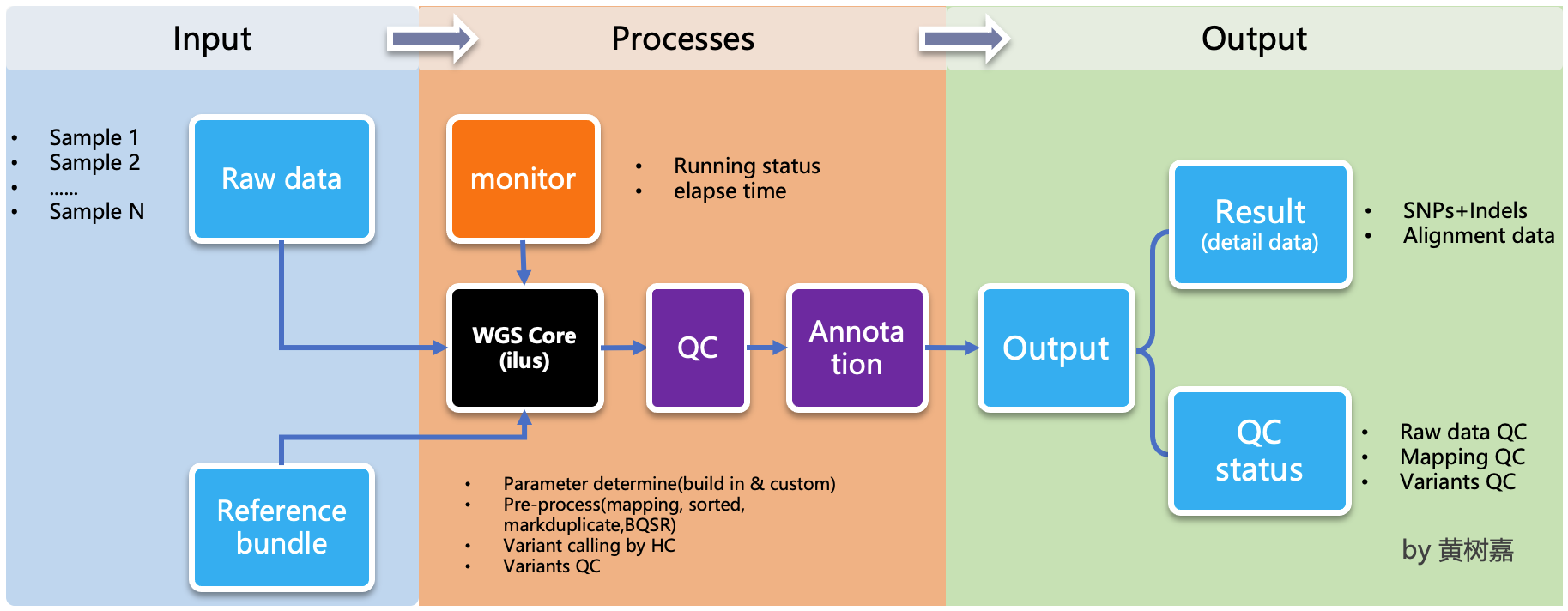
ilus is a pipeline generator, which used to generate WGS/WES analysis pipeline,but ilus can't excute the jobs, which means users needs to submit the jobs by hands, and the processing don't rely on ilus any more,that's why we called it as a semi-automated tools.
ilus has 3 main modelues:
- First、
WGSanalysis module. This module based on GATK Best Practice,usebwa-mem + GATK, the most mainstream way to build an analysis process. It integrates 5 complete processes, including alignment, sorting, and multi-lane merging of the same sample, Markduplicates, HaplotypeCaller gvcf, Joint-calling ,and Variant quality score recalibrator (VQSR). This module also works for WES analysis, Just set the configuration filevariant_calling_intervalparameter to the exon capture interval of WES (detailed below).
By default, the pipeline currently performs the follwing:
- Map reads to reference by `bwa mem`;
- Mark duplicates by `GATK MarkDuplicates`;
- Base quality score recalibration by `GATK BaseRecalibrator` and `GATK ApplyBQSR`;
- Preprocessing quality control by `samtools stats`;
- Calculatiing the base coverage by `bedtools cvg`;
- Variant calling by `GATK HaplotypeCaller`;
- Variants join-calling by `GATK GenotypeGVCFs`;
- Variant quality recalibration by `GATK VariantRecalibrator` and `GATK ApplyVQSR`;
- Annotation by `ensembl VEP`.
-
Second,
genotype-joint-callingmodule. This module is separate from ilus WGS ,in order to call genotype from gvcf directly. Or when you need to complete the WGS/WES data analysis in multiple batches, you can generate gvcf in batches, and finally organize a total gvcf file list, and then use this function to complete the subsequent steps. this can increase the flexibility of the running process. -
Third,
VQSRmodule,also separate from ilus WGS,to help us to quality control on the mutation results.Note that ilus don’t include QC on raw fastq data.
By default, the sequencing data you input is clean data.
ilus does not directly run tasks for the following two considerations:
-
First, avoid making optimizations about task scheduling outside of the core functionality of ilus. Different computing clusters (local and cloud), jobs are scheduled in various ways. If these situations are taken into account, ilus will become bloated and complicated, and it may not be possible to do it. Well, this will cause some people to be unable to use ilus effectively, and even easily lose the focus of ilus in the process of managing task delivery. As a lightweight tool, I designed ilus without the ability to automatically post and run tasks in mind from the start. I hope it can be used as a framework program to generate process scripts that meet your analysis needs strictly based on your input data and configuration file information.
If you want to achieve automatic delivery and monitoring of jobs, ilus It is hoped that it can be implemented in the form of external plugins in the future, but currently these scripts need to be manually delivered.
-
Second, increase flexibility and maneuverability. The processes generated by ilus have a characteristic, that is, they are completely independent of ilus and will no longer rely on any functions of ilus. It doesn't matter if it is completely uninstalled and deleted. In addition, each line in the execution script (shell) generated by ilus can run independently without affecting each other.
The advantage of this is that you can split the script into several sub-scripts according to the characteristics of the computer cluster (if you have few samples or insufficient cluster resources, you can not split them), and then deliver the tasks separately, which can greatly reduce the number of sub-scripts. Speed up the completion of tasks, and, this is currently the only way to deliver ilus tasks in parallel.
How many subscripts to split into depends on your own specific situation. For example, if you have a total of 10 samples, the xxx.step1.bwa.sh comparison script in the first step has a total of 10 comparison commands, and each line is a bwa of a sample.
Since these 10 commands are independent of each other, you can split this step into 10 (or less) subscripts and then manually post these tasks. As for how to split a complete execution script into multiple ones, you can either write your own program or use the yhbatch_slurm_jobs, but note that the program I provide here is based on the slurm system, which may not meet your needs, if you think this the program does not directly meet your needs, you can modify it. Although ilus does not automate the delivery and execution of management tasks, this method can also increase the flexibility and control of process control.
It is very important to monitor the task completion status, especially when there are thousands of samples to be analyzed. This process, if checked manually, would be inefficient and error-prone. ilus has taking this into account, an identifiable completion mark will be added to each task when the process is generated. We only need to check whether each task has this mark (for details, please refer to WGS below for details. example).
Even so, once we have a large number of tasks, it would be too troublesome to manually open the file every time to check whether the corresponding task has been successfully completed. Therefore, I implemented a program in ilus that can be used to check the completion status of the task job. For the specific usage, please refer to the example of WGS below.
ilus is based on Python and supports both Python2.7+ and Python3.7+. The stable version of the code has been released to PyPI. So to use ilus, install it directly through pip the Python package management tool:
$ pip install ilusIn addition to the main program ilus, this command will automatically install other Python packages that ilus depends on. After the installation is complete, execute ilus on the command line. If you can see something similar to the following, then the installation is successful.
$ ilus
usage: ilus [-h] {WGS,genotype-joint-calling,VQSR} ...
ilus: error: too few argumentsBy executing ilus --help you can see three function modules :WGS, genotype-joint-calling and VQSR.
$ ilus --help
usage: ilus [-h] [-v] {WGS,genotype-joint-calling,VQSR,split-jobs,check-jobs} ...
ilus (Version = 1.3.2): A WGS/WES analysis pipeline generator.
optional arguments:
-h, --help show this help message and exit
-v, --version show the version of ilus and exit.
ilus commands:
{WGS,genotype-joint-calling,VQSR,split-jobs,check-jobs}
WGS Create a pipeline for WGS (from FASTQ to genotype VCF).
genotype-joint-calling
Genotype from GVCFs.
VQSR VQSR
split-jobs Split the whole shell into multiple jobs.
check-jobs Check the jobs have finished or not.
That's how you can use ilusBelow, we will introduce how to use these three modules by examples.
The run scripts of the WGS Analysis Pipeline are generated by ilus WGS and are used as follows:
$ ilus WGS --help
usage: ilus WGS [-h] [-n PROJECT_NAME] -C SYSCONF -O OUTDIR [-f] [--use-sentieon] -L FASTQLIST [-c]
[-P WGS_PROCESSES] [-dr]
optional arguments:
-h, --help show this help message and exit
-n PROJECT_NAME, --name PROJECT_NAME
Name of the project. (Default: test)
-C SYSCONF, --conf SYSCONF
YAML configuration file specifying system details.
-O OUTDIR, --outdir OUTDIR
Output directory for results.
-f, --force-overwrite
Force overwrite existing shell scripts and folders.
--use-sentieon Use sentieon (doc: https://support.sentieon.com/manual) to create analysis pipeline.
-L FASTQLIST, --fastqlist FASTQLIST
List of alignment FASTQ files.
-c, --cram Convert BAM to CRAM after BQSR and save alignment file storage.
-P WGS_PROCESSES, --process WGS_PROCESSES
Specify one or more processes (separated by comma) of WGS pipeline. Possible values:
align,markdup,BQSR,gvcf,combineGVCFs,genotype,VQSR
-dr, --dry-run Dry run the pipeline for testing.-C, -L and -O are required parameters, and the rest are optional parameters according to actual needs. The -O parameter is the output directory, if the directory does not exist, ilus will be created automatically. The most important are -C and -L parameters, the former is the configuration file of ilus, without this file ilus cannot generate the analysis process correctly, so it is very important; the latter is Input file, The format of this file has fixed requirements, a total of 5 columns, each column is the necessary information for the process.
Below, I will explain the format of these two files respectively:
The first is the configuration file. We need to write the program path used in the analysis process, the GATK bundle file path, the path of the reference sequence and the parameters corresponding to each key step in the file.
It should be noted that the prefix of the comparison index file of bwa mem should be the same as the prefix of {resources}{reference} of the configuration file, and placed in the same folder. as follows:
/path/human_reference/GRCh38/
|-- human_GRCh38.fa
|-- human_GRCh38.dict
|-- human_GRCh38.fa.amb
|-- human_GRCh38.fa.ann
|-- human_GRCh38.fa.bwt
|-- human_GRCh38.fa.fai
|-- human_GRCh38.fa.pac
`-- human_GRCh38.fa.saThe configuration file should be written in Yaml syntax, here I provide a configuration file template.
In the configuration file, bwa, samtools, bcftools, bedtools, gatk, bgzip and tabix are all necessary bioinformatics software, which need to be installed in advance, and then fill in the path to in the corresponding parameters (as shown in the template). verifyBamID2 is only used to calculate whether there is pollution in the sample, it is not a required parameter, if your configuration file does not have this parameter, it means The process does not calculate the contamination of the sample. If there is, you have to install and download the resource data supporting it. I also tell you where to download the relevant data in the template.
Note that the variant_calling_interval parameter in the configuration file. This is a parameter specifically used to specify the variation detection interval. For example, in the example of the above configuration file, I gave 25 chromosomes from chr1 to chrM, which means to tell the process to perform mutation detection on these 25 chromosomes . If you list only one chromosome in this parameter, or only give a chromosome interval, such as chr1:1-10000, then ilus will also only perform variant detection in the interval you give.
This is a very flexible and useful parameter. The variant_calling_interval interval can be specified arbitrarily. In addition to the assignment method given in my example, you can also assign the interval file path to this parameter. We know that many steps of WGS and WES are exactly the same, and there are only differences in the interval of variant detection ------ WES data is not necessary and cannot be used for variant detection on the whole chromosome, only in the exon capture area.
You only need to put the file of the exon capture area,which in the .bed file format, the example is as follows:
chr1 63697 63697
chr1 101158 101158
chr1 103241 103241
chr1 104108 104108
chr1 185336 185336
chr1 261495 261495
chr1 598862 598862
chr1 601606 601606
chr1 700596 700596
chr1 725086 725086You can also refer to GATK's instructions, here you do not need to manually split them into one by one. It is enough to assign the path of the file to this parameter, and then the process becomes the WES analysis process. This is why ilus is called a WGS and WES analysis pipeline generator.
Also, ilus required public datasets are: gatk bundle and genome reference sequences.
[Note] If the sample size of your project is less than 10, then GATK will not calculate the value of
InbreedingCoeff. In this case,vqsr_optionsin the configuration file does not need to set-an InbreedingCoeff, you can remove it.
Next is the input file specified by the -L parameter. The file contains all the sequencing data information necessary for the WGS/WES analysis process. This file needs to be prepared by you, the format of each column of the file as follows:
-
[1] SAMPLE,Sample name
-
[2] RGID,Read Group,when using
bwa mem's -R parameter -
[3] FASTQ1,Fastq1's file directory
-
[4] FASTQ2,Fastq2's file directory,if it's
Single Endsequencing,the column is replaced with a space -
[5] LANE,fastq‘s
laneidin these file info,
RGIDis the most error-prone, andRGIDmust be set correctly (refer to the following example for the correct way of writing), otherwise the analysis process will go wrong.
In addition, if the sequencing volume of a sample is relatively large, resulting in a sample with multiple lane sequencing data, or the same lane data is split into multiple sub-files, at this time, you do not need to manually analyze these lane data. To merge fastq data, you only need to write the input file according to the sequencing information.
Those data belonging to the same sample, even if the input fastq has been split into thousands of copies, the process will automatically merge after each sub-data is compared and sorted.
Below I give an example of an input file, in which there is a case of the data splitting output of the sample:
#SAMPLE RGID FASTQ1 FASTQ2 LANE
HG002 "@RG\tID:CL100076190_L01\tPL:COMPLETE\tPU:CL100076190_L01_HG002\tLB:CL100076190_L01\tSM:HG002" /path/HG002_NA24385_son/BGISEQ500/BGISEQ500_PCRfree_NA24385_CL100076190_L01_read_1.clean.fq.gz /path/HG002_NA24385_son/BGISEQ500/BGISEQ500_PCRfree_NA24385_CL100076190_L01_read_2.clean.fq.gz CL100076190_L01
HG002 "@RG\tID:CL100076190_L02\tPL:COMPLETE\tPU:CL100076190_L02_HG002\tLB:CL100076190_L02\tSM:HG002" /path/HG002_NA24385_son/BGISEQ500/BGISEQ500_PCRfree_NA24385_CL100076190_L02_read_1.clean.fq.gz /path/HG002_NA24385_son/BGISEQ500/BGISEQ500_PCRfree_NA24385_CL100076190_L02_read_2.clean.fq.gz CL100076190_L02
HG003 "@RG\tID:CL100076246_L01\tPL:COMPLETE\tPU:CL100076246_L01_HG003\tLB:CL100076246_L01\tSM:HG003" /path/HG003_NA24149_father/BGISEQ500/BGISEQ500_PCRfree_NA24149_CL100076246_L01_read_1.clean.fq.gz /path/HG003_NA24149_father/BGISEQ500/BGISEQ500_PCRfree_NA24149_CL100076246_L01_read_2.clean.fq.gz CL100076246_L01
HG003 "@RG\tID:CL100076246_L02\tPL:COMPLETE\tPU:CL100076246_L02_HG003\tLB:CL100076246_L02\tSM:HG003" /path/HG003_NA24149_father/BGISEQ500/BGISEQ500_PCRfree_NA24149_CL100076246_L02_read_1.clean.fq.gz /path/HG003_NA24149_father/BGISEQ500/BGISEQ500_PCRfree_NA24149_CL100076246_L02_read_2.clean.fq.gz CL100076246_L02
HG004 "@RG\tID:CL100076266_L01\tPL:COMPLETE\tPU:CL100076266_L01_HG004\tLB:CL100076266_L01\tSM:HG004" /path/HG004_NA24143_mother/BGISEQ500/BGISEQ500_PCRfree_NA24143_CL100076266_L01_read_1.clean.fq.gz /path/HG004_NA24143_mother/BGISEQ500/BGISEQ500_PCRfree_NA24143_CL100076266_L01_read_2.clean.fq.gz CL100076266_L01
HG004 "@RG\tID:CL100076266_L02\tPL:COMPLETE\tPU:CL100076266_L02_HG004\tLB:CL100076266_L02\tSM:HG004" /path/HG004_NA24143_mother/BGISEQ500/BGISEQ500_PCRfree_NA24143_CL100076266_L02_read_1.clean.fq.gz /path/HG004_NA24143_mother/BGISEQ500/BGISEQ500_PCRfree_NA24143_CL100076266_L02_read_2.clean.fq.gz CL100076266_L02
HG005 "@RG\tID:CL100076244_L01\tPL:COMPLETE\tPU:CL100076244_L01_HG005\tLB:CL100076244_L01\tSM:HG005" /path/HG005_NA24631_son/BGISEQ500/BGISEQ500_PCRfree_NA24631_CL100076244_L01_read_1.clean.fq.gz /path/HG005_NA24631_son/BGISEQ500/BGISEQ500_PCRfree_NA24631_CL100076244_L01_read_2.clean.fq.gz CL100076244_L01The following example illustrates the use and process structure of ilus WGS.
Example 1: Generating a WGS analysis pipeline from scratch
$ ilus WGS -c -n my_wgs -C ilus_sys.yaml -L input.list -O output/This command means that the project (-n) my_wgs generates a WGS analysis process in the output directory output based on the configuration file (-C) ilus_sys.yaml and the input data (-L) input.list . At the same time, the process automatically converts BAM to (-c)CRAM format after completing the analysis. CRAM is more space efficient than BAM, if the -c parameter is not set, the original BAM file is kept.
After the above command is successfully executed, there are a total of 4 folders in the output directory output (as follows):
00.shell/
01.alignment/
02.gvcf/
03.genotype/They are used to store different types of data generated by the process, including:
00.shelltheshellscript collection directory;01.alignmentstores the alignment results in units of samples;02.gvcfstores thegvcfresults of each sample;03.genotypeholds the result of the last variant detection.
There are various execution scripts and log directories of the analysis process in the 00.shell directory:
/00.shell
├── loginfo
│ ├── 01.alignment
│ ├── 01.alignment.e.log.list
│ ├── 01.alignment.o.log.list
│ ├── 02.markdup
│ ├── 02.markdup.e.log.list
│ ├── 02.markdup.o.log.list
│ ├── 03.BQSR
│ ├── 03.BQSR.e.log.list
│ ├── 03.BQSR.o.log.list
│ ├── 04.gvcf
│ ├── 04.gvcf.e.log.list
│ ├── 04.gvcf.o.log.list
│ ├── 05.genotype
│ ├── 05.genotype.e.log.list
│ ├── 05.genotype.o.log.list
│ ├── 06.VQSR
│ ├── 06.VQSR.e.log.list
│ └── 06.VQSR.o.log.list
├── my_wgs.step1.bwa.sh
├── my_wgs.step2.markdup.sh
├── my_wgs.step3.bqsr.sh
├── my_wgs.step4.gvcf.sh
├── my_wgs.step5.genotype.sh
└── my_wgs.step6.VQSR.shWhen submit the task running process, execute it in sequence from step1 to step6. The loginfo/ folder records the running status of all steps of each sample. You can check the .o.log.list log file of each task to get the mark of whether each sample ended successfully.
If successful, you can see a marker like [xxxx] xxxx done at the end of the log file. You can easily know which samples (steps) have been well done, and which ones haven't. This script will help you to output all those unfinished tasks, which is convenient to check for problems and re-execute this part of unfinished tasks. check_jobs_status
The usage is as follows:
$ python check_jobs_status.py loginfo/01.alignment.o.log.list > bwa.unfinish.listif all tasks have done, this list is empty,and print ** All Jobs done **.
The process scripts generated by ilus have a feature, that is, they are completely independent of ilus and will no longer depend on any functions of ilus. At this time, you even use ilus It doesn't matter if you uninstall or delete it. And each line in the execution script (shell) can be run independently.
The advantage of this is that you can split the script into several sub-scripts according to the characteristics of the cluster you are using (if you have few samples or insufficient cluster resources, you can not split them), and then deliver the tasks independently, so it will be more efficient, this is also the only way to submit ilus tasks in parallel.
As for how many subscripts to be split into, it depends on your own needs. For example, if you have a total of 10 samples, there are a total of 10 comparison commands in the xxx.step1.bwa.sh mapping script in the first step. A line is a bwa task of a sample. Since these 10 commands are independent of each other, you can split the step into 10 (or less) subscripts, and then manually post these 10 tasks. As for how to split a complete execution script into multiple ones, you can either write your own program or use the yhbatch_slurm_jobs program provided by ilus , but please note that the program I provide here is based on the slurm system, which may not meet your needs, and you can modify it, if you think This program does not meet your needs.
Example 2: Generate only one/some steps in the WGS process
Sometimes, we do not intend (or do not have to) to complete the WGS process from start to the end. For example, we only want to perform the steps from fastq comparison to gvcf generation, and do not want to perform genotype and VQSR.The -P parameter of ilus can does this:
$ ilus WGS -c -n my_wgs -C ilus_sys.yaml -L input.list -P align,markdup,BQSR,gvcf -O ./outputThis only generates execution scripts from bwa to gvcf, which is useful for projects that need to do analysis in batches. Moreover, the output results of ilus are distinguished by sample units, so in the same output directory, as long as the sample numbers are different, the data of different batches will not overlap each other.
In addition, the -P parameter has another purpose, that is, if a WGS step is wrong and needs to be adjusted, and then re-update the corresponding step, then you can use -P to rerun specific steps. For example, if I need to regenerate the running script of the BQSR step, I can do this:
$ ilus WGS -c -n my_wgs -C ilus_sys.yaml -L input.list -P BQSR -O ./outputHowever, it should be noted that ilus will only keep the total BAM/CRAM file after BQSR for each sample in order to save the project's storage space consumption. Therefore, if you want to re-run BQSR you need to make sure that the BAM file from the previous step of BQSR (ie, markdup) has not been deleted.
If you have been using ilus then you don't need to worry about this problem, because ilus has "atomic properties" when executing tasks, that is to say, only when all processes in the step are successfully completed will the Delete files that are completely unnecessary. Therefore, if the BQSR step does not finish normally, the BAM file of the previous markdup will be preserved.
-P The analysis module specified by the parameter must belong to one or more of「align,markdup,BQSR,gvcf,genotype,VQSR」and be separated by commas.
If we already have gvcf data for each sample, and now use these gvcf to do multi-sample joint variants calling Joint-calling, then you can use
genotype-joint-calling to implement. The specific usage is as follows:
$ ilus genotype-joint-calling --help
usage: ilus genotype-joint-calling [-h] [-n PROJECT_NAME] -C SYSCONF -O OUTDIR [-f] [--use-sentieon] -L GVCFLIST
[--as_pipe_shell_order]
optional arguments:
-h, --help show this help message and exit
-n PROJECT_NAME, --name PROJECT_NAME
Name of the project. (Default: test)
-C SYSCONF, --conf SYSCONF
YAML configuration file specifying system details.
-O OUTDIR, --outdir OUTDIR
Output directory for results.
-f, --force-overwrite
Force overwrite existing shell scripts and folders.
--use-sentieon Use sentieon (doc: https://support.sentieon.com/manual) to create analysis pipeline.
-L GVCFLIST, --gvcflist GVCFLIST
List of GVCF files. One gvcf_file per-row and the format should looks like: [interval
gvcf_file_path]. Column [1] is a symbol which could represent the genome region of the
gvcf_file and column [2] should be the path.
--as_pipe_shell_order
Keep the shell name as the order of `WGS`.-L is the input parameter of ilus genotype-joint-calling, which input a gvcf list file, which consists of two columns, the first column is the corresponding gvcf file The interval or chromosome number, the second column is the path of the gvcf file. Currently ilus requires the gvcf of each sample to be separated by major chromosomes (1-22, X, Y, M), for example:
$ ilus genotype-joint-calling -n my_project -C ilus_sys.yaml -L gvcf.list -O genotype --as_pipe_shell_orderThe format of gvcf.list:
chr1 /path/sample1.chr1.g.vcf.gz
chr1 /paht/sample2.chr1.g.vcf.gz
chr2 /path/sample1.chr2.g.vcf.gz
chr2 /path/sample2.chr2.g.vcf.gz
...
chrM /path/sample1.chrM.g.vcf.gz
chrM /path/sample2.chrM.g.vcf.gzIn this example gvcf.list has only two samples. The parameter --as_pipe_shell_order is optional(the default is not added), its only function is to output the name of the executed script according to the ilus WGS process, maintaining the same order and the same as the WGS process The output directory structure of .
This function is only used to generate execution scripts based on GATK VQSR. If we already have the final mutation detection (VCF format) results, and now we just want to use GATK VQSR to do quality control of this mutation data, then you can use this module, the usage is similar to genotype-joint-calling, as follows:
$ ilus VQSR --help
usage: ilus VQSR [-h] [-n PROJECT_NAME] -C SYSCONF -O OUTDIR [-f] [--use-sentieon] -L VCFLIST
[--as_pipe_shell_order]
optional arguments:
-h, --help show this help message and exit
-n PROJECT_NAME, --name PROJECT_NAME
Name of the project. (Default: test)
-C SYSCONF, --conf SYSCONF
YAML configuration file specifying system details.
-O OUTDIR, --outdir OUTDIR
Output directory for results.
-f, --force-overwrite
Force overwrite existing shell scripts and folders.
--use-sentieon Use sentieon (doc: https://support.sentieon.com/manual) to create analysis pipeline.
-L VCFLIST, --vcflist VCFLIST
VCFs file list. One file per-row.
--as_pipe_shell_order
Keep the shell name as the order of `WGS`.Different from genotype-joint-calling,ilus VQSR input VCF list,and each line is a VCF path,for example:
/path/chr1.vcf.gz
/path/chr2.vcf.gz
...
/path/chrM.vcf.gzOther parameters is the same as genotype-joint-calling. Also, the vcf in the file list does not need to be manually merged in advance, ilus VQSR will do that. A complete example is provided below:
$ ilus VQSR -C ilus_sys.yaml -L vcf.list -O genotype --as_pipe_shell_orderHuang, S., Liu, S., Huang, M. et al. The Born in Guangzhou Cohort Study enables generational genetic discoveries. Nature 626, 565–573 (2024). doi:10.1038/s41586-023-06988-4
-- The End --